Restriction enzymes
are among the best studied examples of DNA binding proteins. In
order to find general patterns in DNA recognition sites, which may
reflect important properties of protein–DNA interaction, we analyse
the binding sites of all known type II restriction endonucleases.
We found a significantly enhanced
G+C
content and discuss three explanations for this phenomenon.
Moreover, we study patterns of nucleotide order in recognition
sites. Our analysis reveals a striking accumulation of adjacent
purines (R) or pyrimidines (Y). We discuss three possible reasons:
RR/YY
dinucleotides are characterized by
These features make RR/YY steps particularly accessible for specific protein–DNA interactions. Finally, we show that the recognition sites of type II restriction enzymes are underrepresented in host genomes and in phage genomes.
|
Nucleic Acids Research 2005 33 (8) : 2726-2733
|
Look at the examples from Kimball's biology pages and translate the sequences into purine (A,G are 1)-pyrimidine (C,T are 0) pattern:
|
Restriction Enzyme |
Source |
Recognition Sequence |
Purine (1)–pyrimidine (0) pattern |
| AluI | Arthrobacter luteus | AG↓CT | 11↓00 |
| HaeIII | Haemophilus aegyptius | GG↓CC | 11↓00 |
| BamHI | Bacillus amyloliquefaciens H | G↓GA TCC | 1↓11 000 |
| HindIII | Haemophilus influenzae | A↓AG CTT | 1↓11 000 |
| EcoRI | Escherichia coli | G↓AA TTC | 1↓11 000 |
FAQs
Q: What can be seen?
A: Restriction enzyme binding sites or recognition sequences are not as diverse as it seems at first sight. Table 1.
Q: How significant is this?
A: The RR/YY motif is statistically much more significant than all other patterns in type II restriction enzyme binding sites. Table 2.
Q: Why is the motif RR/YY preferred?
A: RR/YY dinucleotides are characterized by:
- stronger H-bond donor and acceptor clusters
- specific geometrical properties
- low stacking energy
For details see supplementary information of our article in NAR: supplementary.xls and REBASE
Q: What's about other binding sites?
A: cf. Table 3.
In order to protect themselves, hosts have to methylate the specific binding sites in their own genomes. This happens by methylation of either adenine or cytosine.
There are two different methylation sites in cytosine, but only one in adenine: (n = number of methylations, that reliably prevent DNA cutting)
|
m4 = N4-methylcytosine
n = 146 |
m5 = 5-methylcytosine
n = 1350 |
m6 = 6-methyladenosine
n = 524 |
G and C form 3 H-bonds in complementary base pairing and have a higher binding strength than A and T, which pair with 2 H-bonds. DNA binding proteins better recognize sequences on a bound double strand than on open DNA. One A-T base pair allows for 5 canonical H-bonds between the bases and the recognizing amino acids, whereas the G-C base pair allows for up to 6 H-bonds.
The base decomposition diagramm
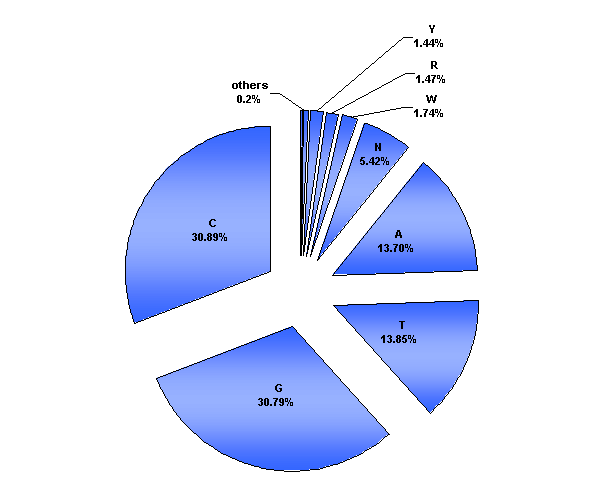
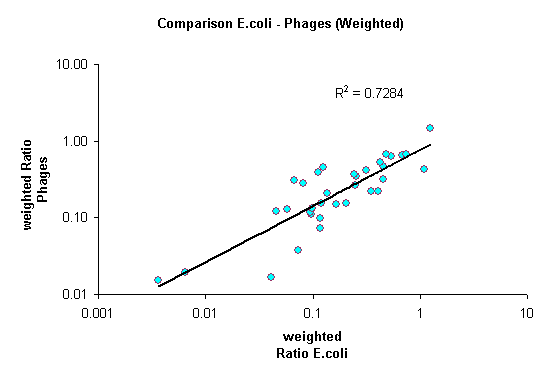
Under-(over)representation in host and phage genomes of E.Coli K12

Additionally, we analysed the genome of E.coli K12 and the known genomes of its phages . All four bases are almost equally abundant in both the E.coli genome and the genomes of its phages. Based on this information we can estimate the expected frequency of any given sequence in a randomized genome. Enrichments of sequences are quantified as the ratio of observed versus expected frequency. In addition we calculated weighted ratios, taking into account the number of different enzymes recognizing the same sequence.
Three findings arise from this analysis:
Your comments are welcome:
mailto:sweta@imb-jena.de
Last update: 26.05.2005